Clinical Lab Quality Control
Clinical Lab Quality Control
The Clinical Lab Quality Control system of CCS, helps a lot the laboratory personnel to maintain the quality of patients test results. The maintenance of quality management system is crucial to a laboratory for providing the correct test results every time. Important elements of a quality management system include Documentation, Standard Operating Procedures (SOPs), Quality Control samples, External Quality Assessment Scheme. Quality Control (QC).
Laboratory quality control (QC) detects, reduces, and corrects deficiencies in a laboratory’s internal analytical process prior to the release of patient results. QC is a measure of precision, or how well the measurement system reproduces the same result over time and under varying operating conditions. Quality Control s not just a procedure. Quality Control IS A SYSTEM. This system consists of:
- Synthetic QC material
- Analytical run
- An understanding of analytical error
- A set of QC rules (algorithms that specify actions)
- A process to follow if the run is deemed to be ‘out of control’
QC Material
Quality control samples are special specimens inserted into the testing process and treated as if they were patient samples. The QC specimens are exposed to the same operating conditions as typical samples. Specimens that are analyzed for QC purposes are known as control materials. To minimize variation, control materials should be available over an extended period of time. Minimal variation should exist and there should be sufficient material from the same lot number or serum pool for one year’s testing.
Both freeze-dried and liquid stabilized control materials are subject to inherent errors which may falsely flag if it is not well understood that there is a problem with the analytical process. There may be instability in certain analytes (for example creatine kinase, bicarbonate) after reconstitution (freeze-dried) or thawing (liquid stable). The act of reconstitution can introduce an error far greater than the inherent error of the rest of the analytical process and there may be introduced contamination from the diluent. Each laboratory should perform stability testing for control material after reconstitution or thawing and for material in long term storage. This should be supported by manufacturer’s documentation.
QC Purpose
The purpose of including quality control samples in analytical runs is to evaluate the reliability of a method by assaying a stable material that resembles patient samples. Quality control is a measure of precision or how well the measurement system reproduces the same result over time and under varying operating conditions.
Biopathologists need to be involved in development of Clinical Lab Quality Control protocols, the selection of quality control materials, long term review of quality control data, and decisions about repeating patient samples after large runs are rejected. These quality control activities play an important part in assuring the quality of laboratory tests.
Controls Usage Procedure
Quality control material is usually run at the beginning of each shift, after an instrument is serviced. In addition, when reagent lots are changed, after calibration, and when patient results seem inappropriate. A quality control scheme must be developed in a way that minimizes reporting of erroneous results. However in must not result in excessive repetition of analytical runs.
The manufacturer should recommend in their product labeling the period of time within which the accuracy and precision of the instruments and reagents are expected to be stable. Each laboratory should use this information to determine their analytical run length, taking into consideration sample stability, reporting intervals of patient results, cost of reanalysis, work flow patterns, and operator characteristics.
Controls run length
The user’s defined run length should not exceed 24 hours or the manufacturer’s recommended run length. Quality control samples must be analyzed at least once during each analytical run. Manufacturers should recommend the nature of quality control specimens and their placement within the run. Random placement of quality control samples yields a more valid estimate of analytical imprecision of patient data than fixed placement and is preferable.
Controls should have target values that are close to medical decision points. Quantitative tests should include a minimum of one control with a target value in the healthy person reference interval and a second control with a target value that would be seen in a sick patient.
Clinical Lab Quality Control Statistical rules
Statistical process control is a set of rules that is used to verify the reliability of patient results based on statistics calculated from the regular testing of control materials. Westgard rules and Levey-Jennings charts are the statistical tools typically used by clinical labs. The most fundamental statistics used by the laboratory are the mean [x] and standard deviation[s]. The mean, or average, is the sum of the control values divided by the total number of values. The standard deviation measures how far, on average, the numbers are from their mean.
Levey–Jennings chart
An example of a Levey–Jennings chart with upper and lower limits of one and two times the standard deviation.
Levey-Jennings chart is a graph that quality control data is plotted on to give a visual indication whether a laboratory test is working well. The distance from the mean is measured in standard deviations (SD). It is named after S. Levey and E. R. Jennings who in suggested the use of Shewhart’s individuals control chart in the clinical laboratory.
On the x-axis the date and time, or more usually the number of the control run, are plotted. A mark is made indicating how far off the actual result was from the mean (which is the expected value for the control). Lines run across the graph at the mean, as well as one, two and sometimes three standard deviations either side of the mean. This makes it easy to see how far off the result was.
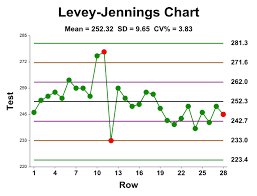
Chart interpretation
Interpretation of quality control data involves both graphical and statistical methods. Quality control data is most easily visualized using a Levey-Jennings control chart. The dates of analyses are plotted along the X-axis and control values are plotted on the Y-axis. The mean and one, two, and three standard deviation limits are also marked on the Y-axis. Inspecting the pattern of plotted points provides a simple way to detect increased random error and shifts or trends in calibration. With a correctly operating system, repeat testing of the same control sample should produce a Gaussian distribution.
That is, approximately 66% of values should fall between the +/- 1 s ranges and be evenly distributed on either side of mean. 95% of values should lie between the +/- 2 s ranges and 99% between the +/- 3 s limits. This means that 1 data point in 20 should fall between either of the 2 s and 3 s limits and 1 data point in 100 will fall outside the 3 s limits in a correctly operating system. In general, the +/- 2 s limits are considered to be warning limits. Values falling between 2 s and 3 s indicates the analysis should be repeated. The +/-3 s limits are rejection limits. When a value falls outside of these limits the analysis should stop, patient results held, and the test system investigated.
Clinical Lab Quality Control Shifts and trends
Reviewing the pattern of points plotted over time is useful in spotting shifts and trends in method calibration. A shift is a sudden change of values from one level of the control chart to another. A common cause of a shift is failure to recalibrate when changing lot numbers of reagents during an analytical run. A trend is a continuous movement of values in one direction over six or more analytical runs. Trends can start on one side of the mean and move across it or can occur entirely on one side of the mean. Trends can be caused by deterioration of reagents, tubing, or light sources. Shifts and trends can occur without loss of precision and can occur together or independently. The occurrence of shifts and trends on the Levey-Jennings control chart is the result of either proportional or constant error.
QC values
When a process is within control, then the values are approximately distributed as follows:
- 68.3% of all the QC values fall within ±1 standard deviation (1s)
- 95.5% of all QC values fall within ±2 standard deviations (2s)
- 4.5% of all data will be outside the ±2s limits when the analytical process is in control
- 99.7% of all QC values are found to be within ±3 standard deviations (3s) of the mean.
Any value outside of ±3s is considered to be associated with a significant error condition. For QC results, any positive or negative deviation away from the calculated mean is defined as random error. There is acceptable (or expected) random error as defined and quantified by the standard deviation (inside the ±3s limits. There is unacceptable (unexpected) random error that is any data point outside the expected population of data (outside the ±3s limits).
Levy-Jennings charts can also demonstrate loss of precision by an increase in the dispersion of points on the control chart. Values can remain within the +/-2 s and 3 s limits, but be unevenly distributed outside of the +/-1 s limits. Random error is present if more than 1 in 20 values fall beyond the +/-2 s limits. By running and evaluating the results of two controls together, trends and shifts can be detected much earlier.
Clinical Lab Quality Control
Westgard rules
Westgard rules are programmed in to automated analyzers to determine when an analytical run should be rejected. These rules need to be applied carefully so that true errors are detected while false rejections are minimized. The rules applied to high volume chemistry and hematology instruments should produce low false rejection rates. A single rule with a large limit, such as the 13s rule, is recommended.
Batch analyzers and manual tests require control samples in each batch. If the test is error prone, the quality control protocol needs to have a high error detection rate. Error detection is improved by increasing the number of controls per run, narrowing control acceptance limits, and using Westgard rules with tighter limits.
Westgard and associates have formulated a series of multi-rules to evaluate paired control runs.
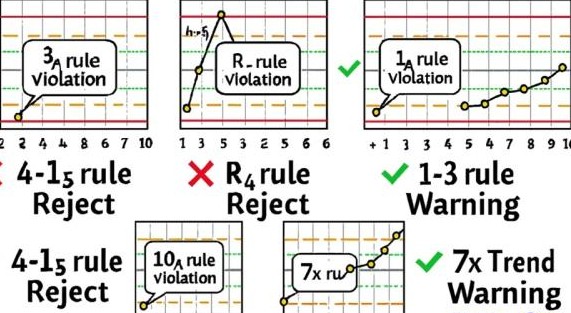
Westgard rules are:
- 12S rule – one control value is outside a 2 standard deviation limit from the mean. Failure of this rule does not always indicate an analytical error has occurred. If the control falls outside of the 2s limit because of a normal Gaussian distribution, the next control result should pass this rule.
- 13S rule – one control value falls outside a 3 standard deviation limit from the mean. This may be the result of random error and should be investigated.
- 22S rule – two consecutive values have fallen outside of the same 2s limit. This rule can apply to a single level during 2 consecutive runs or both levels of control during the same run. Violation suggests systematic error.
- R4S rule – the range between two consecutive values of the same control level is greater than or equal to 4 standard deviations. This rule also applies between control levels. One control is beyond the +2s limit and the other is beyond the -2s limit. Violation suggests random error.
- 41S rule – four consecutive values are have fallen on the same side of the same 1s range. This rule can involve one or both control levels. Violation suggests systematic error.
- 10X rule – ten consecutive values fall on the same side of the mean. This rule can apply within one control level or between control levels. Failure of this rule indicates a shift and the presence of systematic error.
Analytes and accuracy
Analytes with large biological (intra-individual) variation do not require as much analytical accuracy as analytes with small biological variations. One recommendation is that total analytical variation should be less than half the biological variation (see Appendix A for a complete listing of biological variation). For example, the biological variation of fasting triglycerides is ~20%; therefore, analytical variation can be as high as 10% without significantly affecting medical decision making.
Clinical Lab Quality Control Most common problems
Some of the most common problems causing quality control samples to shift are summarized below:
Improper mixing of controls, Change in instrument reaction temperature, Controls left at room temperature too long, Improper reconstitution of control, Instrument sampling problem, Contamination during testing, Vial to vial variation, Concentration of control in error, Instrument reagent delivery problem, Change in reagent lot number (especially with enzymes), Contaminated reagent, Control deteriorated, Instrument malfunction etc.
When problems are identified by Quality Control the following corrective actions can be taken.
Correcive actions
- Check expiration date of the control and the reagent.
- Make sure that the reconstruction of control is proper.
- Retest the control. If the new value is within acceptable limits, record both values and proceed with patient testing. The problem with the first value was probably random error, which is expected in one of every 20 values.
- If the repeat value is still out of range, run a new vial of control. If the new control value is within acceptable limits, record the values and proceed with patient testing. The problem with the first set of controls was probably specimen deterioration.
- Troubleshoot the instrument (check sampling, reagent delivery, mixing, lamp integrity, and reaction temperature).
Recalibrate the method, especially if two or more controls have shifted. - In a shift case, after a new reagent lot number has been introduced, rerun some normal and abnormal patient samples. If patient correlations are good, control shifts are probably acceptable. If they are poor, reagent may be bad.
- Try a new lot number of reagent. If the problem is corrected, check with the manufacturer to find out if anyone else has reported problems.
Understanding of Analytical Error
Effectively this is demonstrated by the emphasis the laboratory places on analytical QC. There should be an appropriately qualified QC coordinator who oversees QA program results and summaries of internal QC results. Evidence should be obvious that these two sets of information are integrated to detect trends and that, based on this, changes are made to calibrations or processes. The head of department should be aware of the state of the QC in the laboratory and of any results which have had to be amended and recalled because of analytical error.
The operational staff should also be able to demonstrate a good understanding of the QC system and what the different types of error are, how they present, what causes them in the analytical system and how to fix them. There should be a documented training program which has evidence that training has been given and received by all staff releasing results.
There must be a system whereby there is demonstrated communication of problems with analytical systems between different shifts. Backup and support for non-expert staff in instrument troubleshooting and QC interpretation should also be evident.
Recording of QC values
It is important to record every quality control value, including those that are out of control. The object of Quality Control is not to produce beautiful control charts by trying to keep all results within +/- 2 SD. Five percent of control values are expected to be out of range. If quality control samples are routinely rerun until they fall within the current control limits and the outliers are not recorded, the acceptable range will become smaller each time they are recalculated. Eventually they will approach zero standard deviation and become useless and unattainable.
When an analytical run is rejected because quality control is out of acceptable limits, it is often necessary to determine if patient results reported between the last acceptable run and the rejected run need to be repeated. This decision should be based on the nature and size of the error. A 5% bias has no clinical significance for most patients, but a 25% bias is unacceptable for nearly all patients. Biases in between these extremes need to be examined on a case by case basis. If analytical errors have clinical significance, then some of the patient specimens should be re-tested. Start retesting with the samples analyzed just before the rejected control samples.
Corrections on test results
Special Clinical Lab Quality Control software routine, supports automatic correction of all patient results of a specific test in a run, if a percentage (positive or negative) bias, is recognised.
For example, suppose all glucose results in a run had a 10% negative bias. A patient with a true blood glucose of 82 mg/dL would have been reported as 74 mg/dL. Both values are normal and the results do not need to be corrected. However, an error of this magnitude would be significant for patient samples near medical decision points.
For instance values below 60 mg/dL, fasting glucose values near 140 mg/dL, and glucose tolerance tests near 180 mg/dL. An acceptable strategy would be to repeat patient samples that were less than 60 or greater than 140 mg/dL. Start retesting with samples analysed just before the quality control failure. Finally repeat testing should be done in reverse chronological order until the new glucose results closely match the original results.
Improved Quality = Increased Productivity at Lower Cost
Laboratory Quality Management
The maintenance of a quality management system like Clinical Lab Quality Control is crucial to a laboratory for providing the correct test results every time. Important elements of a quality management system include:
- Internal quality control (IQC)
- External quality assessment (EQC)
- Proficiency surveillance
- Standard Operating Procedures (SOP’s)
- Documentation
External Quality assessment
This is the evaluation by an outside agency of the performance by a number of laboratories on specially supplied samples. Analysis of performance is retrospective. The objective is to achieve between lab and between method compatibility, but this doesn’t guarantee accuracy unless the specimens have been assayed by a reference lab alongside a reference preparation of known value. Schemes are usually organised on a national or regional basis.
External quality assessment schemes
Aims to analyse the accuracy of the entire testing process from receipt of sample and testing of sample to reporting of results (also known as proficiency testing). Quality in the laboratory is intended to ensure the reliability of the laboratory tests. The objective of quality is to achieve reliable test results by Accuracy and Precision.
Accuracy refers to the closeness of the estimated value to that considered to be true. Accuracy can, as a rule, be checked only by the use of reference materials which have been assayed by reference methods. Precision refers to the responsibility of the result, but a test can be precise without being accurate. Precision can be controlled by replicate tests and by repeated tests on previously measured specimens. And the test result or value which we get should be closer to the previous one.
Inaccuracy and/or imprecision occur as a result of using unreliable standards or reagents, incorrect instrument calibration, or poor technique, eg consistently faulty dilution or the use of a method that gives a reaction that is incomplete or not specific for the test.
Proficiency surveillance
This is concerned with various aspect of laboratory apart from analysis part i.e. this ensures adequate control of the pre and post analytical stages of test. It implies critical supervision of all the aspects of laboratory tests, such as:
- Sample collection
- Labeling
- Delivering
- Storage
- Reading
- Reporting
- Establishment of normal reference values.
- Maintenance and control of apparatus and instruments etc.
- Standardisation
Improve quality – increase productivity
- Eliminating rework
- Save time, save labor, save material e.g. reagent, specimen etc.
- Patient care